
2022年10月19日にFDAはAdvancing Real-World Evidence (RWE) Program(アドバンシング・リアルワールド・エビデンスプログラム)というものを公表しました。

このプログラムは、ごく簡単に説明すると、リアルワールドエビデンス(RWE)を利用して既承認薬の適用拡大や承認後の要求事項の対応によりラベル(≒添付文書)の変更申請につなげるためには、どのようなRWDが必要か?またFDAの許容度はどの程度か?ということが広く理解されるようになるために、FDAとの交渉内容や計画立案の過程を可能な限り公表しながら進めることを前提にした申請制度を設け、スポンサーにはインセンティブを与えることでその制度の利用を促し、結果的に、RWEの規制上の利用の可能性を明らかにし、利用拡大を狙った探索的要素が含まれる制度です。
今回は、このAdvancing RWE Programの解説を見ていきます。
2022年10月20日の連邦官報の通知公表の通り、FDAは、「承認済み医薬品の新しい適応症の承認」や「承認後試験要求(Post-approval study requirements)を満たすこと」を意図したラベル(≒添付文書)の改定をサポートする上での、RWEベース・アプローチの品質と受容性を向上することを模索しているAdvancing Real-World Evidence (RWE) Programを実施する。Advancing RWE Programは、2022年のFDA User Fee Reauthorization Actの一部として組み込まれたPDUFA VIIの下でのFDAのコミットメントを実現するものである
Advancing RWE Programは、プログラムに選ばれたスポンサーに、プロトコールの作成または試験の開始前にFDAスタッフと面談し、医薬品開発におけるRWEの使用について議論する機会を提供する。Advancing RWE Programは、RWEの提案を申請するスポンサーのためのオプションのパスウェイであり、FDAと協議済みの手続きは引き続き利用可能である。
2023年から2027年の間に、FDAの医薬品評価研究センター(CDER)および生物製剤評価研究センター(CBER)はミーティングを実施する予定である。がん領域での申請には、Oncology Center of Excellenceからの参加が予定されている。規制上の決定をサポートすることができるRWEの特性についての知見を高めるため、FDAは本プログラムを通じて議論された試験デザインについて、公開の場(ガイダンスや公開ワークショップなど)で発表する可能性がある。
Advancing RWE Programの目標
Advancing RWE Programは、以下を目的としています。
- 有用性(新しい適応症、集団、用量に関する情報など)に関するラベルのサポート、あるいは承認後試験要求(Post-approval study requirements)を満たすRWEを生成するためのアプローチの特定
- RWEに関する一貫した意思決定と共有学習を促進する規制当局内のプロセスの開発
- Advancing RWE Programで検討された試験デザインについてFDAが公開フォーラムで議論することによる、規制当局の決定をサポートするRWEの特徴についての理解の促進
参加資格
- スポンサーが、Advancing RWE Programのミーティングリクエストに含まれる医薬品のIND(Investigational New Drug)またはpre-IND番号を取得していること
- 提案されたRWEが、有用性(新しい適応症、集団、用量情報など)のラベルをサポートするため、または承認後試験要求(Post-approval study requirements)に対応するための規制上の要件を満たすことを目的としていること
- スポンサーとFDAが、試験デザインに関する情報を一般公開することに合意していること
参加の選択
FDAは、対象となるすべてのRWEの提案に関する申請を歓迎している。しかし、1回の申請サイクルにおける申請件数は限られているため、FDAは、使用に適したデータ(fit-for-use data)、適切な試験デザイン、適切な規制対応を踏まえた実現性に基づき申請を選択する。また、データソース、試験デザイン、解析手法および規制上の適応症の多様性だけでなく、研究の対象疾患や関与しているFDAの部局の多様性も考慮される。
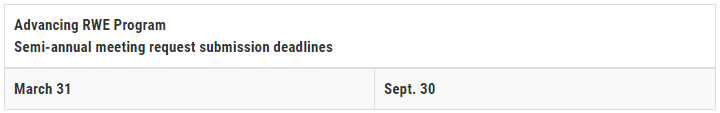
提出期限と手続き
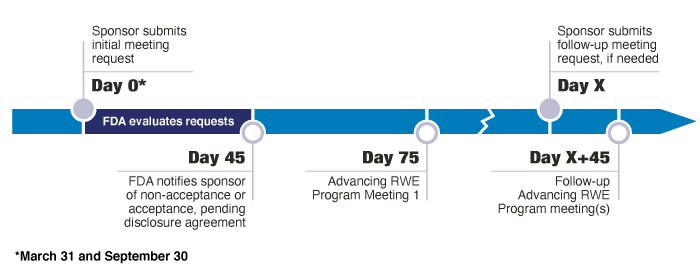
RWE Advancing Programへの初回ミーティングの申請は、3月31日と9月30日の半期ごとに締め切る。 スポンサーは、2027年3月31日まで、Advancing RWE Programに初回のミーティングリクエストをローリング方式で申請することができる。FDAは、各申請の締め切り後、6ヶ月前の申請サイクルで受領したすべてのミーティングの申請について審査する。
FDAは、2023~2024年度には、申請サイクルごとに1~2件の主要会議のリクエストと2件までの代替方法を受け入れ、2025~2027年度には、申請サイクルごとに1~4件の主要会議のリクエストと4件までの代替方法を受け入れる予定である。開示の議論に進むか、会議が拒否されたかは、申請締め切り後約45日後にスポンサーに通知される。本プログラムの一環として認められた各ミーティングリクエストについて、FDAは初回会議と、リクエストがあれば最大3回のフォローアップ会議を実施する。
ミーティングリクエストは、カバーレターの件名に “Advancing RWE Program Meeting Request for CDER” (CDER applications) または “Advancing RWE Program Meeting Request for CBER” (CBER applications)と記載して、関連する申請(例:pre-IND、IND)を電子的に行う必要がある。規制当局への申請書類を電子形式で提供することに関する情報を確認すること。また、Advancing RWE Program のミーティングリクエストが、該当するCDERまたはCBERの申請書に提出されたことを、CDERMedicalPolicy-RealWorldEvidence@fda.hhs.gov 宛に連絡すること。
初回ミーティングリクエストの内容とフォーマット
Advancing RWE Programの初回ミーティングリクエストには、以下の情報を含める必要がある。承認された場合、初回のミーティングリクエストに記載された情報が、初回ミーティングでの議論の基礎となる。要求される情報の範囲は、スポンサーが試験デザインについて最終決定を下す前に、リアルワールド・データ(RWD)を含んだ提案を募集するという目標に沿ったものである。項目#1~8は2ページ以内、項目#9~12は10ページ以内、全体としての上限は12ページとしている。項目#9-12 のトピックのいくつかは関係しないかもしれないし、他は関係するかもしれない。スポンサーは、試験デザイン、データソース、解析、試験実施に関する意思決定に最も関連する情報が何かを決定する必要がある。項目9-12には、潜在的な長所と限界についての議論を含めるべきである。これらの形式および内容の要件に従わないAdvancing RWE Programへの提出物は、それ以上検討されることはない。
- 製品名
- Pre-INDまたはIND番号
- 提案された試験の目的(例:新しいラベルのため、または承認後試験要求(Post-approval study requirements)を満たすため)。
- 提案された適応症(該当する場合)
- 製品開発の簡単な歴史
- RWEアプローチを取る理由
- 提案されている試験デザインの簡単な概要
- スポンサーが開示できないと考える試験デザインの要素(該当する場合、除外する根拠とともに)
- 試験デザインの属性:目的、デザイン(例:プラグマティックな要素を含む無作為化試験、外部対照試験、観察コホート研究)と概略図;適格基準;関心のある共変量;主要評価項目及び重要な副次的評価項目;関心のある治療、比較対象及び併用治療
- 潜在的なデータソース:カテゴリー(例:電子カルテ、医事請求、レジストリ、その他)と説明;データの信頼性(reliability)と関連性(relevance);主要なデータ要素のバリデーション、タイミング、完全性;他のデータ源とのリンケージ;追加的なデータ収集
- 予想される解析計画:おおよそのサンプル数;主要及び副次的な評価項目に関する解析計画;交絡因子へのアプローチ;追跡期間の定義;同時発生イベント、データの欠損または誤分類、多重性の扱い
- その他の考慮事項:試験デザインと実施方法の事前規定;患者レベルデータの利用可能性とFDAによるアクセス、被験者保護への取り組み
情報開示に関する合意
提出期限後45日以内に、FDAは会議リクエストをレビューし、開示協議に進む主要および代替リクエストを選択し、スポンサーにその状況を通知する。
イノベーションを促進し、異なるタイプのデータソースや試験デザインの許容性についての明確化のために、Advancing RWE Programを通じて開発された主要なデザイン要素は、研究対象の医薬品がまだFDAによって承認されていない場合や市販後試験が完了していない場合も含めて、FDAによってケーススタディとして公表される場合がある(例:ガイダンスや公開ワークショップで)。FDAは、RWEの理解とその潜在的な規制上の利用に関する要素に焦点を当てる意向である。スポンサーのプログラムが公開議論の焦点となることが分かっている場合には、可能であれば、FDAは事前にスポンサーに通知する。
FDAは、Advancing RWE Programの下で初回のミーティングを許可する前に、FDAとスポンサーは、FDAが公に開示する情報について合意しなければならない。FDAは、スポンサーとの開示合意において、データソースおよび試験デザインについて説明する以下のカテゴリーの情報を、該当する場合、含めることを意図している:
- RWEの提案された利用方法
- 承認済み製品のラベル変更のサポート:新しい適応症;新しい集団への使用の拡大;用量・用法・投与経路の変更;その他のラベルの変更
- 承認後試験要求(Post-approval study requirements)のサポートまたは充足
- 対象となる疾患・病態
- リアルワールドのデータソース
- データソースの分類(例:電子カルテ、医事請求、レジストリ、その他)及び簡単な説明
- データの信頼性(データ収集と保証のプロセスを含む)
- 取り組んでいるリサーチクエスチョンに対するデータの関連性(Relevance)
- 主要なデータ要素のタイミングと完全性
- データの完全性の保証
- 主要なデータ要素に関連するバリデーション
- 他のデータソースや追加的なデータ収集とのリンケージ
- 患者レベルのデータや原資料へのFDAによるアクセス方法
- 研究デザインの特徴:
- デザインの種類(例:RWDを組み込んだ無作為化試験、RWDを組み込んだ外部対照試験、観察コホート)
- 研究スキーム(曝露および追跡期間など)
- サンプルサイズおよび集団に関する一般的な説明
- 研究対象集団のサンプルサイズと一般的な説明
- 対照薬/比較薬の選択
- 研究のエンドポイント
- 関心のあるestimand
- 研究モニタリングの計画
- 無作為化および盲検化(該当する場合)
- 解析計画
- 帰無仮説と対立仮説
- バイアスの評価とコントロールの方法
- 主要評価項目および主要な副次的評価項目に対する統計的検定
- 重要なサブグループ解析
- 欠測や誤分類の取り扱い方法
- 多重性に対処するためのアプローチ
- Advancing RWE Programに関連して、提案されたRWEについて申請者とFDAとの間で行われるやりとりの中で生じる上記のいずれかの変更または修正
開示対象からの除外
FDAは、スポンサー名、利害関係のある治療法(製品名や分子構造など)、試験適格基準の完全な説明、または患者レベルのデータに関する詳細を共有する予定はない。
このプログラムの情報開示の側面は、すべての参加当事者に一貫して適用されることが重要である。もしスポンサーが、自分たちのプログラムには特有の情報開示に関する考慮事項があると考えた場合、スポンサーは開示不可と考える情報を特定し、その情報を差し控える根拠を提供する必要がある。Advancing RWE Programへの参加は、情報開示に関する合意を含め、スポンサーの自由意思に基づくものとする。情報開示を希望しないスポンサーは、既存のルートを通じて規制当局の意見を求めることができる。
フォローアップミーティングのリクエストの内容とフォーマット
Advancing RWE Programの一環としての初回ミーティングのリクエストが認められたスポンサーで、合計3回までのフォローアップミーティングを希望する場合には、フォローアップミーティングリクエストおよび情報パッケージを電子的に提出すること。カバーレターの件名に、”Advancing RWE Program Follow-up Meeting Request for CDER”(CDER申請の場合)または”Advancing RWE Program Follow-up Meeting Request for CBER”(CBER申請の場合)を含めること。また、Advancing RWE Programフォローアップミーティングリクエストが該当するCDERまたはCBERに申請されたことを、CDERMedicalPolicy-RealWorldEvidence@fda.hhs.gov 宛に連絡すること。
フォローアップミーティング・パッケージは、以下の情報を含んでいる必要がある:
- 製品名
- Pre-INDまたはIND番号
- 提案の主な変更点、新しい情報、または前回の会議以降の決定事項
- フォローアップミーティングでFDAと議論するための質問リストと、その質問の必要性または背景の説明
初回およびフォローアップのAdvancing RWE Programのミーティングから30日以内に、ミーティングの要約は依頼者に送付される。
以上、Advancing RWE Programの制度についての説明でした。
さすがFDAと言いますか、実験的な要素のある、とても面白い制度です。
新医薬品の申請の場合であれば機密情報の開示を恐れ、たとえインセンティブがあっても製薬企業が乗りにくい制度になってしまうと思いますが、適用拡大や製販後であればちょうど良いバランスのような感じがしました。

